
Hot topic 4: Biomolecular simulations
Franca Fraternali, Randall Centre for Cell and Molecular Biophysics, King’s College London, United Kingdom
Chris Oostenbrink, University of Natural Resources and Life Sciences, Vienna, Institute of Molecular Modeling and Simulation, Austria
Molecular simulation has become an important tool in the molecular sciences, which offers insight at a level often unattainable experimentally. With the broad availability of computational resources, more and more scientists with a background in wet-lab experiments enter the field and perform simulations of complex molecular systems. On this day, we will cover key topics in the area of Biomolecular Modelling and Simulation. The first part involves a general overview on modeling and simulation of biological systems, the second part a more in depth analysis of how these methods can be used to compute freeenergy differences and to address allosteric effects.
Part I: General overview on modeling and simulation on biological systems
In the first part, we will discuss the basics of molecular dynamics simulations. Starting from the link between a molecular structure and the (potential) energy of a system, we will discuss various algorithm to modify the structure in physically meaningful ways. This will lead to the definition of ensembles and the computational tools to generate ensembles of complex biomolecular systems. We can subsequently use the statistical mechanical ensembles to calculate averages of molecular properties which may be compared to experimental data directly. Next, we will mention methods to define ensembles at different thermodynamic state points. Finally, we will discuss possibilities to use experimental observations as boundary conditions for molecular simulations, in order to e.g. refine molecular structures. Using NMR parameters like NOE distance restraints or 3J-coupling constants as examples, the various ways of restraining the molecular structure will be discussed.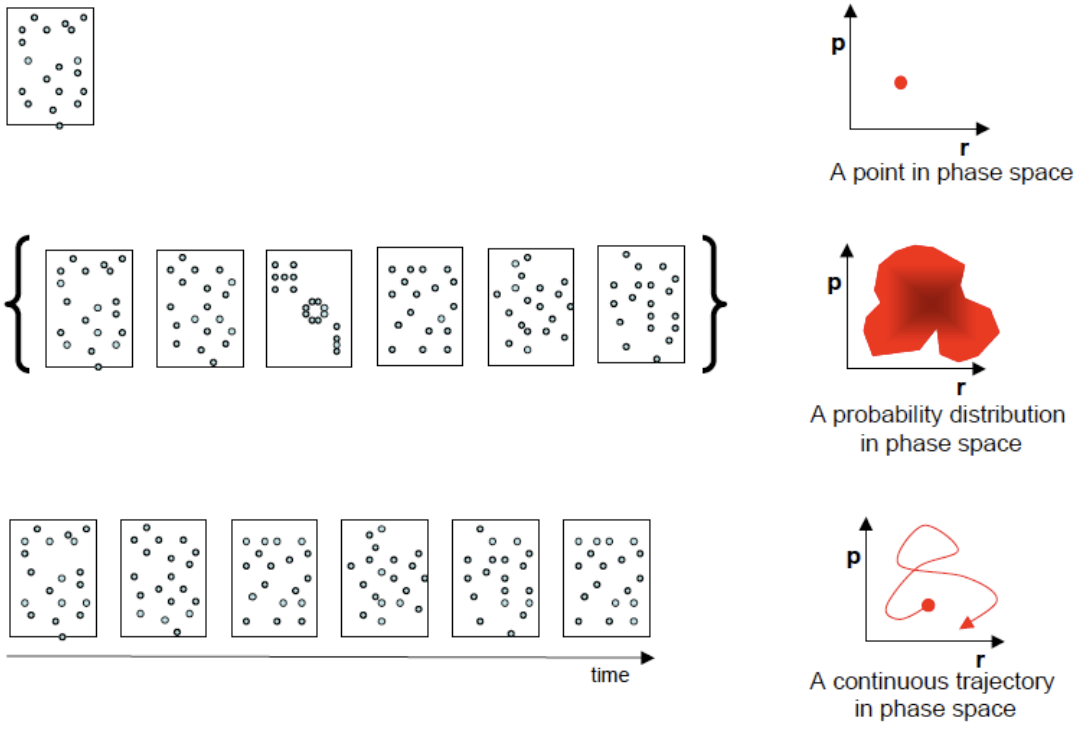
Modelling protein structures and analysing simulated trajectories
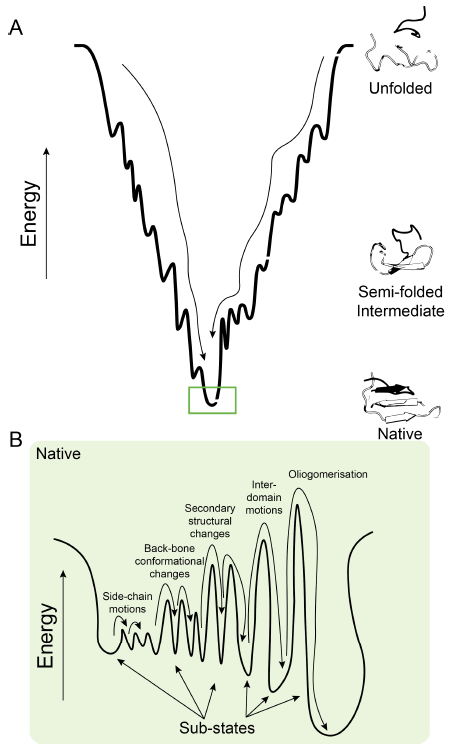
The importance of accurately modelling proteins will be illustrated by reviewing current protein prediction techniques and discussing their relevance in the context of molecular simulations. Static model representations of proteins are so prevalent that it is easy to misconstrue these dynamic machines for rigid aperiodic crystals. Proteins attain their native structure via the process of protein folding, where they reach the native state through a funnel-shaped path of energetic intermediates. Folded proteins can adopt a variety of different structural and energetic conformations that form ensembles of isoenergetic ground states, referred as conformational sub-states. Determination of sub-states can be facilitated by bioinformatic tools. We will show the derivation of a structural alphabet from encoding all high-resolution structures in the Protein Data Bank. This alphabet, whose letters are a set of canonical backbone fragments, can be applied to monitor conformational transitions in simulated trajectories.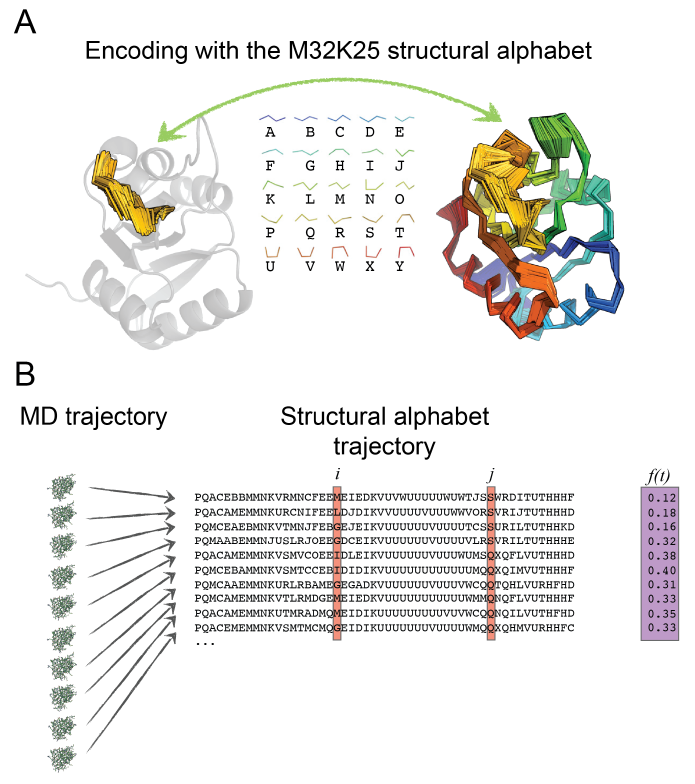
The following videos are from paper: Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface.
References
- Maria M. Reif et al., Testing of the GROMOS Force-Field Parameter Set 54A8: Structural Properties of Electrolyte Solutions, Lipid Bilayers, and Proteins. Chem Theory Comput. 2013 Feb 12; 9(2): 1247–1264. Published online 2013 Jan 2. doi: 10.1021/ct300874c
- W. F. van Gunsteren et al., Deriving structural information from experimentally measured data on biomolecules. Angewandte Chemie International Edition 55.52 (2016): 15990-16010. doi: 10.1002/anie.201601828
- Alessandro Pandini et al., GSATools: analysis of allosteric communication and functional local motions using a structural alphabet. Bioinformatics. 2013 Aug 15; 29(16): 2053–2055. Published online 2013 Jun 5. doi: 10.1093/bioinformatics/btt326
- Jeroen Claus et al., Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. eLife. 2018; 7: e32271. Published online 2018 May 1. doi: 10.7554/eLife.32271
Part 2: Free energies and allostery
Case studies – Allosterically regulated proteins: PKM2 (mammalian pyruvate kinase); NtrC; GPCRs
The free energy forms the driving force of any molecular process. Intrinsically containing both enthalpic and entropic contributions, an accurate estimation of free energies is possible from statistical mechanical principles. This lecture will use the free energy of ligand binding as a representative example for which such calculations may be performed. We will first introduce the various actors and their enthalpic or entropic contribution to the binding affinity and subsequently focus on the alchemical free energy methods that can be used to calculate the free energies. Real case examples from our own work will be used to demonstrate the use of the methods.
Analysis of allosteric signalling in protein simulations
We will illustrate techniques to extract correlated motions from sampled conformational states. In particular, network representations of proteins will be introduced together with techniques illustrating how these reduced representations can be queried to extract allosteric signalling. We will ask the question of how to identify specific protein residues that propagate the signal of allosteric ligand binding. This is a mandatory step for designing targeted experiments that can validate predictions about the signalling mechanism. We will illustrate successful examples of this strategy from our own work.
References
- Manuela Maurer et al., Comparison of free‐energy methods using a tripeptide‐water model system. Comput Chem. 2018 Oct 5; 39(26): 2226–2242. Published online 2018 Oct 2. doi: 10.1002/jcc.25537
- Jan Walther Perthold et al., Simulation of Reversible Protein–Protein Binding and Calculation of Binding Free Energies Using Perturbed Distance Restraints. Chem Theory Comput. 2017 Nov 14; 13(11): 5697–5708. Published online 2017 Sep 12. doi: 10.1021/acs.jctc.7b00706
- Alessandro Pandini et. al., Detection of allosteric signal transmission by information-theoretic analysis of protein dynamics. FASEB J. 2012 Feb; 26(2): 868–881. doi: 10.1096/fj.11-190868
- A.B. Nørholm et al., Thermodynamic characterization of new positive allosteric modulators binding to the glutamate receptor A2 ligand-binding domain: combining experimental and computational methods unravels differences in driving forces J. Chem. Inf. Model 54 (2014) 3404−3416 doi: 10.1021/ci500559b
Part 3: Hands-on session – Extracting dynamical signals in practice
We will discuss examples proposed by the students in the context of free energy methods, their advantages and pitfalls. The students will be guided through tutorials of online methods to measure flexibility and effects of mutations on protein stability. We will introduce our stand-alone software package AlloHubMat (Allosteric Hub prediction using Matrices), utilised to predict allosteric hub fragments from the network of dynamically correlated motions. A step-by-step tutorial has been devised based on examples from our work. We will stimulate a discussion about the advantages and disadvantages of coarse-grained versus atomistic models in assessing residue flexibility and correlated motions in protein structures.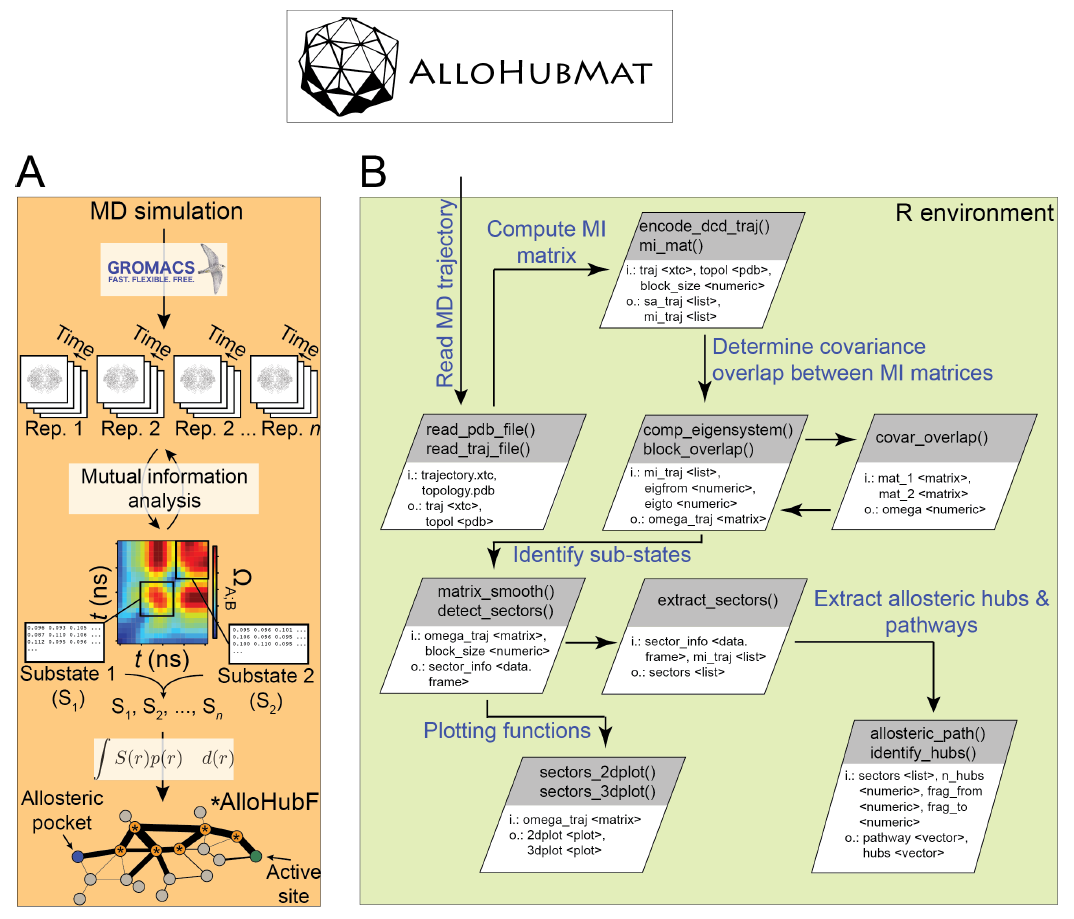
References
- Macpherson JA et al., Functional cross-talk between allosteric effects of activating and inhibiting ligands underlies PKM2 regulation. bioRxiv (2018), https://www.biorxiv.org/content/early/2018/07/26/378133
- Guarnera E et al., AlloSigMA: allosteric signaling and mutation analysis server. Bioinformatics. 2017 Dec 15;33(24):3996-3998. doi: 10.1093/bioinformatics/btx430. PubMed PMID: 29106449.