
Hot topic 2: Cryo-Electron Microscopy (cryo-EM)
Jürgen Plitzko, Max Planck Institute of Biochemistry, Department of Molecular Structural Biology, Germany
Carrie Bernecky, Institute of Science and Technology Austria, Klosterneuburg, Austria
In this Hot Topic session, we will offer lectures and a guide on how to do cryo-electron microscopy (Cryo-EM) as of today. We explain and show how cryo-EM has advanced, what is now state-of-the-art and what can be expected in the future. The aim of this session is to build the basis to understand and perform cryo-EM in both theory and practice.
Cryo-EM on vitrified specimens is a formidable tool to visualize macromolecules with the transmission electron microscope (TEM) in three dimensions (3D) and at near-atomic resolution. Three different techniques are the backbone of 3D characterization with the TEM: electron crystallography (cryo-EC), single particle EM (cryo-EM), and electron tomography (cryo-ET).
Electron crystallography has been primarily used for the analysis of two-dimensional (2D) protein crystals. Although it was the only method to obtain high-resolution structures for a very long time, it suffered from the fact that only a few proteins could be “forced” into such two-dimensional crystalline arrays and thus prevented a broad application of this method. Recently, however, microcrystal electron diffraction (MicroED) has been resurrected to investigate single 3D macromolecular crystals with a thickness between 0.1- 1 µm at high resolution.
Single particle cryo-EM (SPA) has traditionally been used to provide insights into the morphology of large purified protein complexes that have resisted crystallization, albeit at much lower resolutions than crystallography. However, recent technological advances in sample preparation, computation and, in particular, instrumentation have enabled researchers to solve macromolecular structures with near atomic resolution using cryo-EM. As a consequence of this ‘resolution revolution’ cryo-EM single particle analysis has become the most versatile method for structural biology. Whilst crystallography is strongly restricted by the conformation entrapped in the crystals, single particle analysis is able to cover the entire spectrum of conformations including those relevant for cellular conditions. Once the remaining bottleneck in controlling the unpredictable behavior of proteins during sample preparation is solved, it is likely that cryo-EM particle analysis will gradually replace X-ray crystallography to become the primary method for protein structure determination.
Cryo-electron tomography has a lot in common with single particle cryo-EM. However, there are decisive differences and one is quite apparent: tomography allows imaging of molecules in cells quasi in vivo, while in SPA molecules are investigated separate and apart from their cellular hosts and their interacting partners, thus in vitro. Tomography therefore can be used to image the cellular proteome and to reveal the network of interactions between the many molecular species that give rise to cellular function. However, the main challenge of cryo-ET is the sample thickness, necessitating appropriate sample preparation protocols to obtain fully vitreous and homogeneous thin electron-transparent (< 300 nm) slices from cells or tissues. While the resolution of tomograms is modest, the averaging of subvolumes containing the same type of particles (analogous to single particle analysis) allows the improvement of resolution, an approach called subtomogram averaging (STA). Subtomogram averages of sufficiently frequent complexes typically reach resolutions in the range of 15 Å (and in favorable cases below 10 Å). Subtomogram averaging can be more demanding than SPA due to the lower particle count, their higher variability, and the higher background and noise levels. Nevertheless, the target structures can be localized in their native environment, so that many different states can be detected including the presence of labile or transient interaction partners.
Part 1: An Introduction to cryo-EM
This introductory lecture is aimed at the ever-increasing circle of scientists that wish to become familiar with electron microscopy and is supposed to provide an overview of the current state of the art in cryo-EM investigation and preparation. It is important to note that the preparation techniques must be specially adapted to the peculiar physical characteristics of the sample itself and the electron microscopic image formation. Therefore, we will give a general introduction of the transmission electron microscope and the most important preparation techniques for cryo-EM and describe their pros and cons. Since this introduction is aimed primarily at the newcomer rather than the expert, it is only about the most important laws of electron optics that are necessary to understand the operation and the practical use of the electron microscope. For this purpose, practical questions such as parallel illumination, the correction of astigmatism, the correction of coma, the use of direct electron detectors, phase plates and their alignment are addressed in more detail. Besides, the interaction of the electrons with the specimen is described, how contrast is generated, how images are formed, and how radiation damage can be minimized by low-dose techniques.
References
- R. F. Thompson et al., An introduction to sample preparation and imaging by cryo-electron microscopy for structural biology. Methods. 2016 May 1;100:3-15. doi: 10.1016/j.ymeth.2016.02.017. Epub 2016 Feb 28.
- J. Arnold et al., Site-Specific Cryo-focused Ion Beam Sample Preparation Guided by 3D Correlative Microscopy. Biophys J. 2016 Feb 23;110(4):860-9. doi: 10.1016/j.bpj.2015.10.053. Epub 2016 Jan 5.
Part 2: Journey from a sample to the 3D structure
Part 2 of this hot topic session is focused on the computational pipeline for SPA and introduces the principles that govern the relationship between a three-dimensional object and its projections (i.e. the projection theorem). It also explains the basis for recovering the object from a finite set of projections and discusses the relationship between signal-to-noise-ratio (SNR), the number of projections, and the object size to the resolution with which it can be retrieved. The next topic is the most difficult part of SPA: the determination of particle orientations, since each particle image corresponds to a different molecular view with a different and unknown orientation. Orientations can be assigned with an ab initio model (deduced from the particle images) or with reference to an existing 3D template obtained from data that directly delivers orientation angle values (e.g. from random conical tilt or subtomogram averages). We will also address the need for 2D and 3D classification to account for the heterogeneity of the sample (i.e. different conformations or different macromolecular species). Therefore, techniques such as multivariate data analysis, principal component analysis and correspondence analysis are presented and explained.
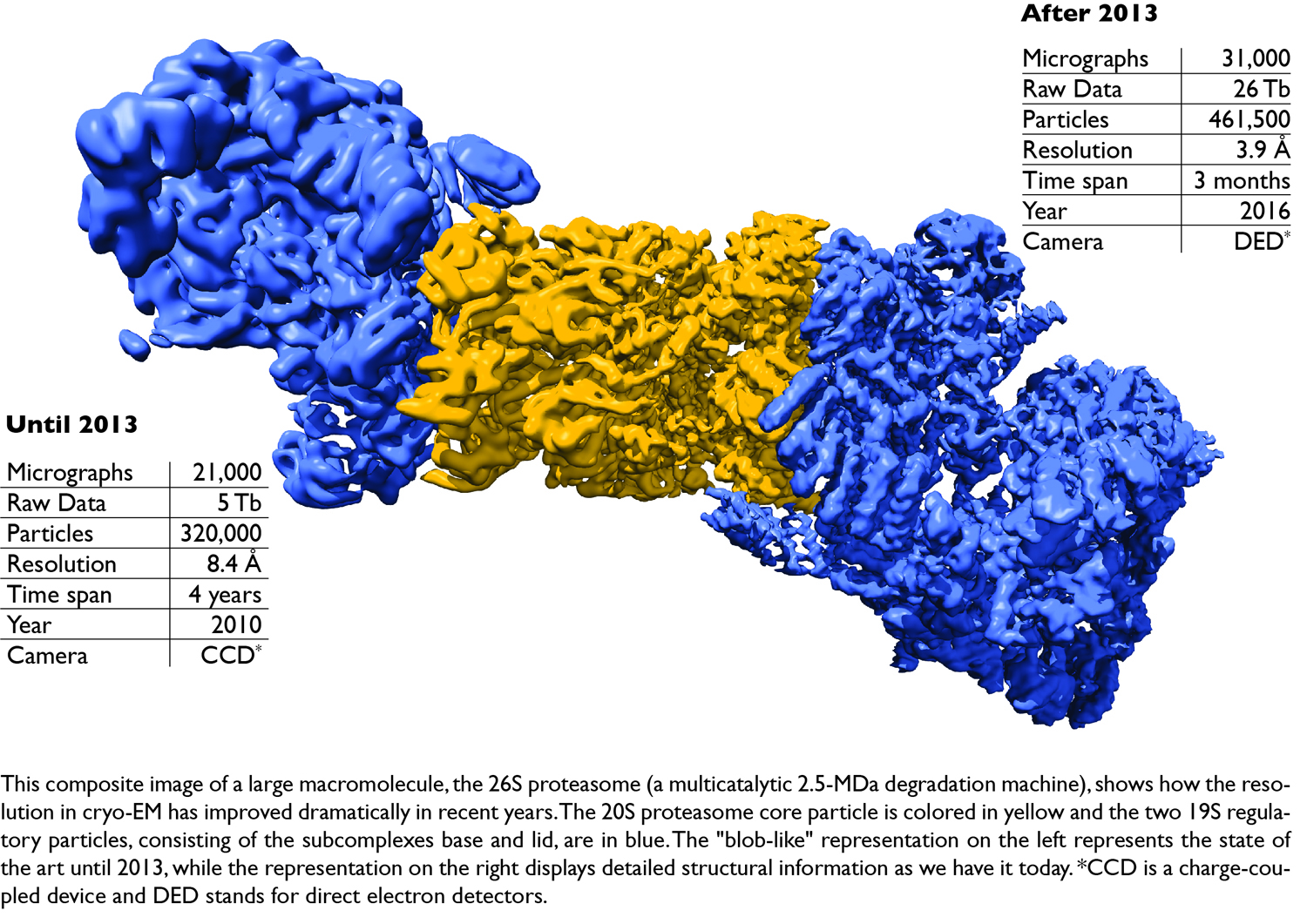
Computational tools are an important part of the cryo-EM structure determination process that follows data acquisition, and therefore practical questions are also addressed to avoid model bias, to obtain structures from smaller difficult-to-crystallize proteins such as G-protein-coupled receptors by means of phase plates, and to correctly dock atomic resolution structures in EM maps with lower resolution. In this image processing tutorial for cryo-EM, we will show how single particle analysis is performed on macromolecular complexes ranging from large megadalton machines (such as the 26S proteasome) to medium-sized (RNA polymerase II) to very demanding small proteins (smaller than 200 kDa, such as G protein coupled receptors; GPCRs).
References
- A. Schweitzer et al., Structure of the human 26S proteasome at a resolution of 3.9 Å. Proc Natl Acad Sci U S A. 2016 Jul 12;113(28):7816-21. doi: 10.1073/pnas.1608050113. Epub 2016 Jun 24.
- C. Bernecky et al., Structure of transcribing mammalian RNA polymerase II. Nature. 2016 Jan 28;529(7587):551-4. doi: 10.1038/nature16482. Epub 2016 Jan 20.
- Y. L. Liang et al., Phase-plate cryo-EM structure of a class B GPCR-G-protein complex. Nature. 2017 Jun 1;546(7656):118-123. doi: 10.1038/nature22327. Epub 2017 Apr 24.
Part 3: What’s next?
Single particle cryo-EM is now fairly mature in terms of data acquisition and image processing software. The remaining bottleneck is controlling the unpredictable behavior of proteins in sample preparation. In order to obtain cryo-EM grids of sufficient quality for high-resolution single particle analysis, careful optimization of many variables is required. Common challenges to overcome are sample-related limitations (i.e. buffer compositions), particle distribution (e.g. obtaining the correct concentration), preferred orientation, and poor reproducibility of the grid production process. Therefore, in our last session, we will discuss new approaches to sample preparation for SPA, such as affinity grids and the spot-it-on method.
Despite the hurdles we have to overcome when preparing samples for SPA, it is obvious that the future of structural biology lies in situ and is made possible by cryo-electron tomography (cryo-ET). This is because cellular functions arise from the interactions between the many molecular species that inhabit cells. Cryo-ET of cells is an extremely rich source of structural information depending on cell physiology; however, access to this information is clearly underdeveloped. Therefore, new approaches are urgently needed to make better use of the information content of tomograms, that we will explore in our last session. While the first part of this session belongs to the future, the last part of this session will be interactive and will address questions raised during the sessions and during the course of this workshop on cryo-EM. In addition, we will debate, discuss and provide feedback on everything that has been requested and suggested by the participants.
References
- A.J Noble et al., Reducing effects of particle adsorption to the air-water interface in cryo-EM. Nat Methods. 2018 Oct;15(10):793-795. doi: 10.1038/s41592-018-0139-3. Epub 2018 Sep 24.
- Mahamid et al., Visualizing the molecular sociology at the HeLa cell nuclear periphery. Science. 2016 Feb 26;351(6276):969-72. doi: 10.1126/science.aad8857.