prof. Elspeth Garman 
The University of Oxford, Department of Biochemistry,
Oxford, UK
Background:
The aim is to flash-cool crystals in order to reduce the rate of radiation damage during exposure to X-rays. Protein crystals contain up to 80% solvent (water) and water can form ice crystals upon freezing. Ice crystals will 1) perturb the crystalline lattice due to the 7% volume increase of ice as compared to water and 2) give raise to diffraction during exposure to X-rays, which will parasite the diffraction pattern generated by the protein crystals. To avoid the formation of ice crystals, the crystal is usually soaked in a cryo-protectant. Cryoprotectants lower the freezing point of water (just as ‘anti-freeze’ is added to the cooling water of a car in winter) and more importantly, they also slow down the kinetics in the solution by increasing the mean free path of water molecules meeting other water molecules to form ice. With luck, crystals might grow from crystallization solutions that already have cryo-protectant properties. In this case, the crystal can be transferred directly from the crystallization droplet to the nitrogen gas stream (or liquid nitrogen), a procedure called “fish-and-cool”.
In practice, a crystal usually has to be soaked for a few seconds in a solution of the same composition as your crystallization solution complemented with the cryo-protectant. (ideally with the water in the solution replaced by the cryo-protectant, rather than just diluting your mother liquor with the cryo-protectant). However, as such a solution is different from the crystallization solution, the crystal might not tolerate the new environment and crack (osmotic shock) or lose its sharp edges(solubility change). The ideal soaking solution has the water of the crystallization buffer replaced by the cryoprotectant, rather than being made by just diluting the mother liquor with the cryoprotectant.
The object is 1) to find the nature of cryo-protectant tolerated best by the crystal and 2) to find the suitable concentration of cryo-protectant. This concentration should be low enough such that the cryo-solution is as similar as possible to the crystallization solution and does not harm the crystal, and high enough such that ice crystals do not form.
If the crystal does not diffract following cryo-cooling, it may be that it never diffracted in the first place, so testing it at room temperature should be the next step. Then it will be clear whether it was the cryo-cooling protocol that destroyed the diffraction, or whether further crystallization trials are required.
Demonstration/Practical
Demonstration A: Mounting for room temperature testing
Room temperature mounting equipment will be demonstrated using the MiTeGen system: a metal ‘top hat’ with a loop attached which has two different diameters of stem over which the plastic sleeve fits snuggly, and a black plastic sleeve guide (‘aligner’) for threading polyamide sleeve onto the pin with containing the looped crystal so that the crystal is not deposited on the inside of the sleeve instead of staying in the loop. The size of the loop is matched to the size of the crystal before ‘fishing’ so that the crystal cannot move around in the liquid across the loop during the RT data collection.
The polyamide sleeve is sealed at one end and contains a small volume of the crystallization buffer which has been previously pipetted into the end of the sleeve. This prevents crystal dehydration during RT data collection.
Each student will practice threading the plastic sleeve onto the pin using the sleeve guide.
Demonstration B: Optimum crystal fishing strategies
‘Soup spoon’ versus ‘minimize surrounding film’ crystal fishing techniques will be shown.
Demonstration C: Storage of crystals in pucks
The crystal holding pucks and tools for loading them will be explained.
Demonstration D: Keep crystal moving under liquid nitrogen
When flash-cooling a crystal into a Dewar of liquid next the microscope, the Dewar lid should be used to flap away the cold gas layer above the liquid nitrogen before plunging the crystal in. Also, very importantly, immediately after plunging, gently move the crystal on the wand around in the liquid nitrogen in order to leave behind the insulating bubbles of gas that form around the crystal/loop. This ensures a fast cooling rate. The Dewar should be kept as full as possible to avoid having a layer of warmer gas above the surface which gives a temperature gradient through which the crystal must travel to reach the liquid, and thus it cools more slowly.
Demonstration E: Cryovial transfer to canes
If any using canes and vials to store crystals, there will be a demonstration of a good way to handle the vials.
With gloves on (both insulating and plastic lab gloves) hold your palm UPWARDS and FLAT with a gap between the index finger and the other 3 digits. Hold the vial in pincer movement keeping fingers FLAT (people tend to curl their fingers up and then this method doesn’t work well).
Each person using canes should practice loading and unloading a vial in this way from a warm cane.
N.B. NEVER has the 2-liter tall tin Dewar full of liquid nitrogen on the bench. It should ALWAYS be on the floor, ideally in a simple stand to stop it from being kicked over.
Below are two exercises that will be outlined and participants will carry out some of the steps. There will not be a goniometer or liquid nitrogen at HTCC5, but improvisation will portray the ideas, and then all the steps can be carried out at home!
Exercise 1: Fishing crystals from Linbro plates and flash-cooling them in a 100 K nitrogen stream
Method:
- Under the microscope look over the Linbro-plate plate and find a drop with well-formed crystals. The crystals should be good for data collection: single, and clean edges.
- Under the microscope look at different cryo-loops (Hampton or MiTeGen) and select one of the sizes comparable to the size of the crystal you decided to fish (This is very important because the following steps have to be carried out quickly and there is no time to waste selecting a suitable loop).
- Place the cryo-loop on the goniometer head and make sure that the center of the loop aligns with the trajectory of the X-ray beam (the center of the loop in the center of the camera). Adjust the distance if necessary.
- Verify that the cryo-stream is well aligned with the center of the cryo-loop. IMPORTANT.
- Take off the coverslip with the drop you selected in step 1 (you may need to use a scalpel blade to lever up the coverslip) and place it on the cover of the Linbro plate (obviously upside-down).
- Pipette 2 ml of the cryo-solution into a nearby empty well or, if there are only 2 or 3 crystals in your well, place it next to the droplet containing the crystals.
Note: if there are a lot of crystals in the well, DO NOT put the resolution on top of them! The crystals might degrade in the time it takes to fish them all, and the resolution may dehydrate the crystals, shrinking the unit cell of the crystals left in there for longer times. This will make merging data collected from different crystals at the data analysis stage more challenging.
- Clip the cryo-loop you selected in step 2 on the crystal wand, fish a crystal, bathe it for a few seconds in the cryo-solution, fish it again, and very importantly, BLOCK THE STREAM. Put the cryo-loop onto the goniometer head. Once it is there safely, unblock the stream as quickly as possible (i.e. remove the blocker, usually a strip of cardboard, very fast to ‘flash-cool’ the crystals).
Note: DO NOT refocus the microscope to check that the crystal is in the loop, as the crystal will dehydrate during that time and the mosaicity may increase.
- Do not forget to put the coverslip back over the reservoir to prevent the crystals from dehydration.
Exercise 2: Fishing crystals from 96-well plates and flash-cool them in liquid nitrogen
Method:
- Under the microscope look over the 96-well plate and find a drop with well-formed crystals. The crystals should be suitable for data collection: single, and clean edges. Write down the well number.
- Under the microscope look at different cryo-loops (Hampton or MiTeGen) and select one of the sizes comparable to the size of the crystal you decided to fish.
- If using cryo-vials, with the help of the tweezers transfer the empty vial of the cryo-loop within the sample holder basket placed in the Dewar containing liquid nitrogen.
- With the plate under the microscope, use the scalpel to cut the tape, which covers the drop of choice.
- If the crystal of choice is attached to other crystals (see a drop on the very right in the scheme below), try to carefully separate the crystals with an acupuncture needle. If there is skin over the crystals, use two needles, one in each hand. Hold the skin down with one needle (keep your hand still) and chop around the skin with the other needle.
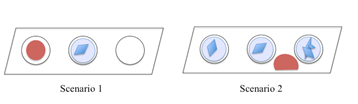
- Pipette 1 ml of the cryo-solution next to the droplet containing crystals. Scenario 1: If only one of the crystallization wells contains a crystallization drop (blue), use one of the empty wells to add the cryo-solution (red). Scenario 2: If all crystallization wells contain crystallization drops, put the cryo-solution on the shelf between the crystallization drops. Be careful and make sure that the drop does not move towards the crystallization well and join with the crystallization drop (see point 6 above).
- Clips the cryo-loop you selected in step 2 onto the crystal wand, fish a crystal, bath it for a few seconds in the cryo-solution and transfers it quickly into the liquid nitrogen. KEEP THE CRYSTAL MOVING GENTLY after plunging it, in order to leave behind gaseous bubbles of nitrogen which insulate the crystal from the cold liquid and which can prevent efficient cooling. Then once the bubbling has ceased, place it within the empty vial placed under liquid nitrogen. Feed the crystal pin straight into a puck by looking directly down the wand held perpendicularly to the plane of the puck (i.e. put your face over the Dewar while wearing safety glasses to line up the pin carefully not to lose the crystal on the inside of the puck).
- If the crystal want be fished because it is stuck to the surface of the crystallization well, try first to lift it by waving the cryo-loop near it in the crystallization well. If this procedure fails, scratch the well near the crystal with the acupuncture needle to stress the plastic. This sometimes releases the crystal. If this does not work, try to detach it directly with the acupuncture needle and repeat step 7.
- Do not forget to seal the open well with sealing tape.
Questions:
- How can you find the best suitable cryo-protectant?
- Why is it important to put the crystal in the center of the cryo-stream?
- Did you find it more difficult to fish crystals from a Linbro plate or from a 96-well plate?
Some additional background explanations:
How can we cool the crystal to 100 K without destroying the ordered lattice? Small changes in procedure can have a significant impact on the resulting resolution of the data and thus impact the quality of the biological information obtained. The crystal must be cooled so fast that the solvent in the intermolecular cavities becomes an amorphous glassy solid, rather than as ordered ice crystallites. The latter disrupts the internal ordered lattice when they form, since their volume is greater than that of liquid, and also give powder diffraction rings that obscure reflections at particular resolutions (3 intense powder rings between 3.5 and 3.9 Å). Thus it is often necessary to add an organic solvent, a ‘cryo-protectant’ such as glycerol, ethylene glycol, or light polyethylene glycols to the crystal buffer and soak the crystal in it prior to swiftly plunging it into liquid nitrogen (77 K).
Thus optimizing the cryoprotection step is important for the final outcome, and typically the experimenter takes the crystallization buffer and dilutes it with the cryo-protectant. Sometimes the crystal (imagine YOU are the crystal!) does not take kindly to this treatment, since the protein solubility might increase and then the sharp edges of the crystal start to become rounded, or at the other extreme, the osmotic pressure of this cryoprotectant buffer might be much larger/smaller than prior to soaking, and small ‘crazy paving’ cracks will appear on the crystal surface. If either of these problems is encountered, it is worth making up the cryoprotectant buffer by replacing the water in the original crystallization buffer with the cryoprotectant agent (i.e. making up the latter at double concentration and then adding water and cryoprotectant agent to reproduce the original crystallization buffer concentrations). In problematic cases, this method has often given very good results. Also, if the osmotic shock is the issue, sequential in situ soaking in increasing concentrations of cryoprotectant buffer can have a huge positive impact (e.g., the resolution of subtype N6 influenza neuraminidase crystals, which required 40% glycerol for optimum protection, routinely increased from 3.2 Å to 1.6 Å resolution after using this protocol).
For two reasons, harvesting just one crystal at a time for soaking in the cryo-buffer is recommended, rather than pipetting the buffer onto a whole drop containing many crystals.
Firstly, many cryo-protectant agents cause dehydration and shrinkage of the unit cell, and since fishing them takes time for each crystal, they will spend varying times soaking and thus have a range of unit cells, which will give increased systematic errors if data from different crystals are to be merged. Secondly, many crystals will remain stable in the cryoprotectant buffer for a limited time, but will then start to degrade, so it is unwise to leave them in it for too long while cooling the other crystals.
The next step is to ‘fish’ the crystal into a cryo-loop under a microscope, first checking that there is no skin over the drop and that it is not stuck to the bottom of the well, by stirring the drop with a tool (acupuncture needle, cat’s whisker, etc). To minimize the amount of liquid around the crystal (and thus minimize the background-ray scattering) the loop can be brought out of the drop with its plane perpendicular to the drop surface. To avoid ice forming in the liquid nitrogen, the Dewar should be kept as full as possible and have an easily removable lid on top of it during ‘fishing’, and the nitrogen should be regularly renewed.
If the cryo-cooled crystal doesn’t diffract, testing at RT can be very worthwhile as then it can be determined whether the crystal lattice was destroyed by the cryo-cooling protocol which should then be altered (soak time, soak temperature, and cryoprotectant) or whether it was never well ordered in the first place, and new crystallization trials are then required.
Crystals for data collection at 100 K at synchrotrons are typically now cooled into the pucks that fit straight into the beamline mounting robot, and then shipped to the synchrotron in special transport Dewars. These Dewars should be allowed to dry out after every trip, in order to disperse water vapor that has condensed and been trapped inside when they are opened and closed.