HT2: Processing
L4 (Lecture): Refinement and Model Building
Rob Nicholls
MRC Laboratory of Molecular Biology
Cambridge Biomedical Campus
Cambridge, UK
Refinement of atomic structural models against crystallographic experimental data is an integral part of crystal structure analysis. Since crystallographic observations are intensities of the corresponding structure factors, and there is no direct way of observing the phases, most crystallographic computations revolve around recovering the lost phases. Hence refinement in general has two purposes: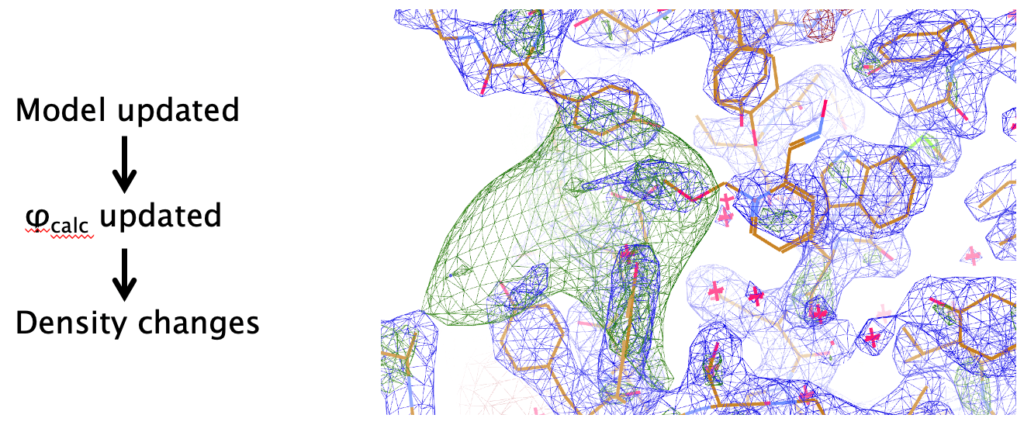
- To derive as accurate atomic structural models as possible, and
- To improve model phases thus generating the best possible electron density maps.
Although the main aim of macromolecular crystallography (MX) is to derive accurate atomic models in order to answer specific biological questions, the importance of improving phases and the resulting electron density maps should not be underestimated. Such maps help in automatic and manual model building, affecting the quality of final atomic models.
Effects such as crystal mosaicity and disorder lead to poor diffraction quality and weak intensities, resulting in only low-resolution data being available. Such high-resolution information loss worsens the observation-to-parameter ratio, which results in unstable refinement, overfitting, and ultimately an unreliable model. Consequently, regularisers are used in order to stabilise MX refinement, and ensure consistency between the derived model and available prior knowledge. Using the Bayesian framework, such regularisers are implemented in the form of restraints, which are often referred to as “geometry terms”. Regularisers are typically used at all resolutions, although more may be required at lower resolutions in order to achieve an acceptable effective parameter-to-observation ratio. Indeed, challenges that are encountered during model parameterization and refinement may vary depending on the high-resolution limit of the collected diffraction data.
Restraints representing chemical information are commonly used at all resolutions (e.g. bond and angle restraints), which help local structure adopt chemically reasonable conformations. At medium resolution, TLS, local NCS, and B-value restraints may be used. At lower resolutions, additional supplementary “external restraints” may be needed in order to encourage consistency with models of homologous structures, formation of hydrogen bonding networks, nucleotide base pairing and stacking. Notably, jelly-body restraints stabilise MX refinement without injecting externally derived information.
When external restraints are used, an anharmonic penalty function is used to control robustness to any outliers caused by inconsistencies between data and prior information. These external restraints and robust estimation procedures are also useful during model building, where necessary to increase real-space refinement convergence radius and stability.
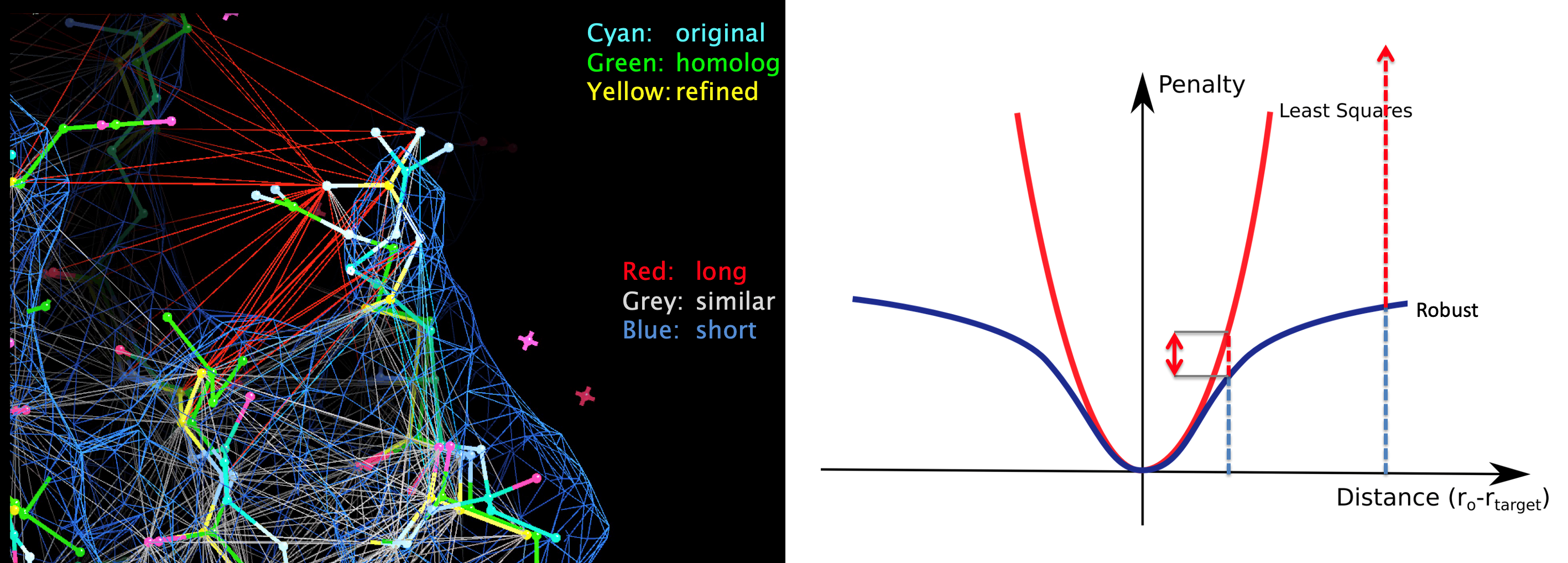
Where several datasets and models are available for a macromolecule, restraints can facilitate information transfer between structures, potentially improving refinement and thus resultant model quality. Pragmatically, determining suitability of reference structures and refinement parameters can be challenging; the automated pipeline LORESTR facilitates this process by trialing multiple protocols.
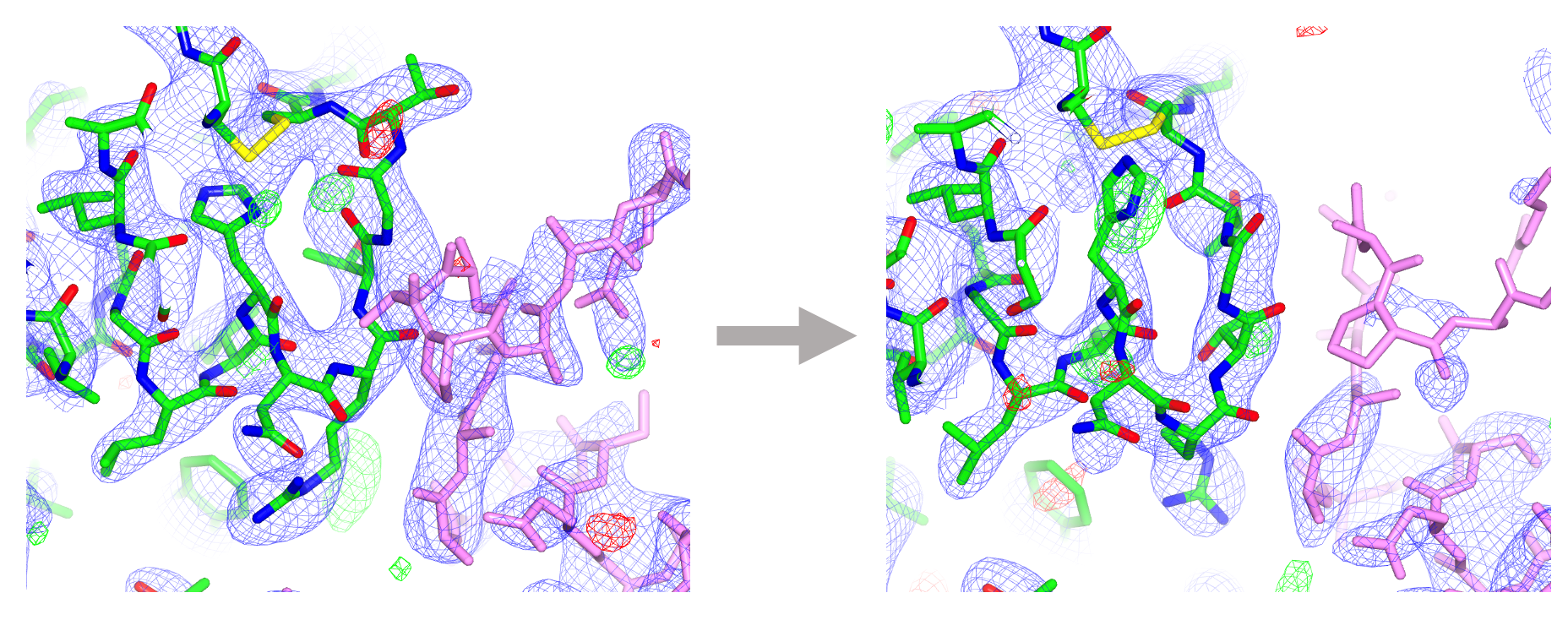
Additional consideration must be given in cases where a model contains a novel ligand/compound for which a description does not yet exist in the CCP4 Monomer Library, or if a compound is covalently linked to the macromolecule (post-translation modifications). In such cases, a bespoke restraint dictionary corresponding to the compound and/or linkage is required in order to that the model maintains sensible chemistry and acceptable geometry. Such dictionaries can be generated using AceDRG. Following refinement in REFMAC5, visualisation and validation tools in Coot facilitate manual inspection, critique and improvement of the protein-ligand complex model.
In this session, we will discuss techniques to facilitate the refinement of high-quality MX models, including more challenging cases where only low-resolution data are available. We shall focus on implementations within the CCP4 suite, specifically using the software tools: REFMAC5, Coot, AceDRG, ProSMART, LibG and LORESTR.
Low-resolution MX refinement with REFMAC5, ProSMART, LibG & LORESTR:
- Nicholls et al. (2017) Low Resolution Refinement of Atomic Models Against Crystallographic Data. Protein Crystallography, 565-93.
- Nicholls et al. (2013) Recent Advances in Low Resolution Refinement Tools in REFMAC5. Methods for Bio. Xtallography, 231-58.
- Nicholls et al. (2012) Low Resolution Refinement Tools in REFMAC5. Acta Cryst. D68, 404-17.
Tools for ligand fitting & validation with CCP4/Coot:
- Nicholls (2017) Ligand fitting with CCP4. Acta Cryst. D73, 158-70.
- Emsley (2017) Tools for ligand validation in Coot. Acta Cryst. D73, 203-10.
- Debreczeni & Emsley (2012) Handling ligands with Coot. Acta Cryst. D68, 425-30.
Modelling covalent linkages:
- Nicholls et al. (2021) Modelling covalent linkages in CCP4. Acta Cryst D77, 712-26
- Nicholls et al. (2021) The missing link: covalent linkages in structural models. Acta Cryst D77, 727-45.
Primary software references:
REFMAC5: Murshudov et al. (2011) REFMAC5 for the refinement of macromolecular crystal structures. Acta Cryst. D67, 355-67.
Coot: Emsley et al. (2010) Features and development of Coot. Acta Cryst. D66, 486-501.
AceDRG: Long et al. (2017) AceDRG: a stereochemical description generator for ligands. Acta Cryst. D66, 486-501.
ProSMART: Nicholls et al. (2014) Conformation-Independent Structural Comparison of macromolecules with ProSMART. Acta Cryst. D70, 2487-99.
LibG: Brown et al. (2015) Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Cryst. D71, 136-53.
LORESTR: Kovalevskiy et al. (2016) Automated refinement of macromolecular structures at low resolution using prior information. Acta Cryst. D72, 1149-61.
Additional reading relevant to refinement with REFMAC5 and associated tools:
Effect of Twinning on R-factors:
- Murshudov GN (2011) Some properties of Crystallographic Reliability Index – Rfactor: Effect of Twinning. & Comp. Math., 10, 250-61.
Cooperative utilisation of information from MX and NMR:
- Kovalevskiy et al. (2018) Overview of refinement procedures within REFMAC5: Utilising Data from Different Sources. Acta Cryst. D74, 215-27.
- Carlon et al. (2016) How to tackle protein structural data from solution and solid state: An integrated approach. Progress in nuclear magnetic resonance spectroscopy. 92, 54-70.
Tools for cryo-EM model fitting & refinement:
- Casanal et al. (2020) Current developments in Coot for macromolecular model building of electron cryo-microscopy and crystallographic data. Protein Science 29(4), 1055-64.
- Nicholls et al. (2018) Current approaches for the fitting and refinement of atomic models into cryo-EM maps using CCP-EM. Acta Cryst. D74, 492-505.
- Murshudov (2016) Refinement of atomic structures against cryo-EM maps. Methods in Enzymology, 277-305.
- Brown et al. (2015) Tools for macromolecular model building and refinement into electron cryo-microscopy reconstructions. Acta Cryst. D71, 136-53.