HT2: Processing
The underlying idea of structure determination by means of X-ray diffraction on crystals is based on the fact that diffraction intensities represent square amplitudes of structure factors, or Fourier Transform coefficients, of the crystallised structure. If both structure factors amplitudes and phases were known from the experiment, then the electron density could be easily obtained as their inverse Fourier Transform. However, phase information is not recorded in the experiment and, therefore, needs to be elicited by other means. This is known as the Phase Problem in Crystallography. In tutorials T2-T3 we will learn how to solve the Phase Problem with CCP4. Solved Phase problem yields initial approximation to electron density and atomic structure, which should be further improved with iterative Model Building and Refinement. This will be considered in L4, T4 and T5.
T2: Tutorial: Automatic and Fundamental Molecular Replacement
Eugene Krissinel, Maria Fando

Science and Technology Facilities Council, CCP4 core group
Oxford, UK
From MTZ and sequence to a built and refined model via MR route with CCP4 Cloud; using AlphaFold-2 models, AlphaFold-2 database, and Structure Prediction task in CCP4 Cloud
Molecular Replacement (MR) is the most popular method to solve the Phase Problem nowadays. MR is based on the assumption that target structure has a highly similar structural homologue, which may be found by, e.g. matching target’s sequence to sequences of known structures. In this method, a sufficiently close molecular model is positioned (i.e., rotated and translated) in crystal coordinates such that calculated structure amplitudes reproduce experimental results in the best possible way. Then, calculated phases are considered as first approximations to real phases, which allow for subsequent model building, refinement and phase improvement.
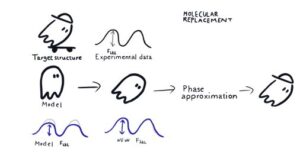
For the success of Molecular Replacement, the MR model should be sufficiently similar to the target structure. In this tutorial, we will discuss how to find and prepare suitable MR Search Models (including working with Alphafold2 predicted models) for use in molecular replacement programs (Phaser and Molrep). We will also cover the basic principles of MR and the software available in CCP4 for each step of MR, including both fundamental tools and automatic pipelines that combine all needful steps and can deliver phased structure starting from experimental observation and sequence – or even without the later.
T3: Tutorial: Automatic and Fundamental Experimental Phasing
Eugene Krissinel, Maria Fando
Science and Technology Facilities Council, CCP4 core group
Oxford, UK
From MTZ with anomalous signal and sequence to a built and refined model via EP route with CCP4 Cloud
MR is based on the assumption that a close structure homolog does exist. So, for solving a structure we need a similar structure solved. How was the first ever structure solved then? In principle, structure can be found from reflection data by so-called Direct Methods, based on phase relationships between structure factors. Direct Methods were developed by Herbert Hauptman and Jerome Karle, for which they won the 1985 Nobel Prize in Chemistry (https://www.nobelprize.org/prizes/chemistry/1985/summary/). Direct Methods have been implemented in several software packages (e.g., Shelx).
As appears, for purely technical reasons, such as natural presence of noise in experimental data, Direct Methods can be used only for limited-size structures, depending on the resolution (usually limited by 10-100 atoms; as a maximum, ~1000 atom structures have been reported solved by the method). Therefore, Direct Methods are not generally suitable for determining macromolecular structures. However, if we could have diffraction data from a small subset of atoms in the crystal, then we can find their positions with Direct Methods, calculate their phases and use them for the estimation of the overall structure’s phases. A set of phasing techniques, based on this idea, is called Experimental Phasing.
In this workshop, we will focus on the most popular Single-wavelength Anomalous Diffraction (SAD) phasing. SAD approach exploits the anomalous scattering from a set of selected atoms in the crystal, which can be identified in diffraction images (such a set is commonly called as heavy-atom substructure). We will discuss what an anomalous signal is and how we can estimate its properties for a given anomalous scatterer, such as Se, S, Zn, Mg and others.
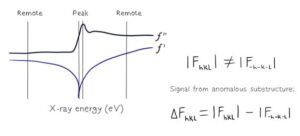
Next, we will learn how to use various software available in CCP4 to find the heavy-atom substructure. Since diffraction from a substructure is not distinguishable from diffraction of its inverse, the software produces coordinates and phases for both of them. Such sets of substructures and phases are commonly called “original” and “inverted” hands.
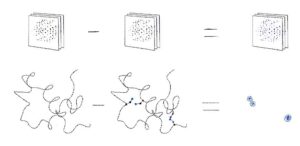
However, only one hand gives the correct protein phases. We will see how the correct hand can be identified with CCP4 software, and solve a structure with SAD phasing using prepared data containing a signal from anomalous scattering. We will learn how this can be done in CCP4 using both fundamental programs or sophisticated pipelines. As a result we shall get the phased structure from just reflection data and sequence – or give you a protein backbone if sequence is not known.